








《水产品的检验PPT课件下载(共28张)》是由用户上传到老师板报网,本为文库资料,大小为1.28 MB,总共有29页,格式为ppt。授权方式为VIP用户下载,成为老师板报网VIP用户马上下载此课件。文件完整,下载后可编辑修改。
- 文库资料
- 29页
- 1.28 MB
- VIP模板
- ppt
- 数字产品不支持退货
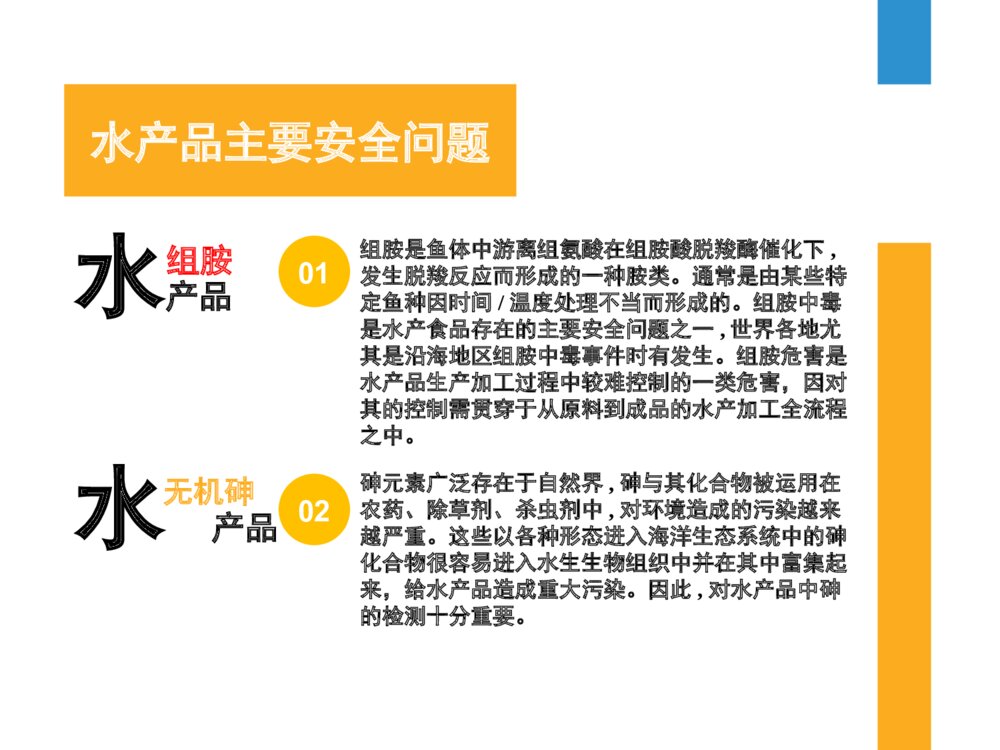
水产品的检验的安全现状水产品水产品质量安全现状我国是水产品生产大国, 连续多年居世界首位.水产食品营养丰富、味道鲜美, 并具有低脂肪、高蛋白、营养平衡性好的特点, 深受人们的喜爱。由于水产品易受海、淡水域环境污染的影响和药物在水产养殖中的大量使用, 因此水产品安全性倍受国内外的关注。水产品和人们的生活健康密切相关,所以需要国家地方和行业制定相关标准来保障水产品的质量安全。我国正在积极开展渔业标准化体系建设,已经制定了相关的标准。水产品主要安全问题水产品组胺组胺是鱼体中游离组氨酸在组胺酸脱羧酶催化下,发生脱羧反应而形成的一种胺类。通常是由某些特定鱼种因时间/温度处理不当而形成的。组胺中毒是水产食品存在的主要安全问题之一,世界各地尤其是沿海地区组胺中毒事件时有发生。组胺危害是水产品生产加工过程中较难控制的一类危害,因对其的控制需贯穿于从原料到成品的水产加工全流程之中。01水 产品无机砷砷元素广泛存在于自然界,砷与其化合物被运用在农药、除草剂、杀虫剂中,对环境造成的污染越来越严重。这些以各种形态进入海洋生态系统中的砷化合物很容易进入水生生物组织中并在其中富集起来,给水产品造成重大污染。因此,对水产品中砷的检测十分重要。02水产品主要安全问题水 产品甲基汞汞(hg)及其化合物是环境中广泛存在的一类持久性污染物,对人类健康造成很大的威胁,汞及其化合物曾大量用于杀虫剂、杀菌剂、化妆品等领域。工业废水、废气、含汞化合物中的汞会通过人类活动进入到外界环境中,且会在食物链中逐渐富集。目前每年仍有大量的各种形态的汞排放到外界环境中,造成很大的污染环境中的汞污染物经过大气沉降、水的流动等方式最终进入到地表水中,对水产品养殖和生存环境造成很大的污染,养殖环境中各种形态汞化合物很容易被水产品吸收和富集,严重影响水产品质量,由于其在水产品中残留时间长,并有可能通过食物链进行传递,进而对人类产生巨大的危害。因此,对水产品中汞残留进行检测势在必行。0102的测定法组胺组胺测定法1、分光光度法(GB/T5009.45)012、液相色谱法(GB5009.208—2016)023、高效液相色谱-紫外检测法(参考文献:徐丽,刘琳,王宗奇,孟慧琴,张雪琰.高效液相色谱-紫外检测法测定水产品中组胺含量[J].食品安全质量检测学报,2014,5(12):3790-3794)03第一法:分光光度法•(1)原理:样品中的组胺用正戊醇提取后,在弱碱性条件下与重氮盐发生反应生产橙色的偶氮化合物,与标准系列比较定量。•(2)方法说明:适用于水产品等含有组胺的食品。•本方法的检出限为50mg/kg。•(3)样品前处理:秤取一定量绞碎并混匀的样品,加入三氯乙酸溶液浸泡以提取组胺并沉淀蛋白质,过滤。吸取适量滤液,用氢氧化钠溶液调制碱性,组胺游离,用正戊醇萃取游离组胺。吸取适量正戊醇提取液,加盐酸至酸性,组胺成为盐酸盐而易溶于水溶液,反萃取至盐酸溶液中,合并盐酸提取液并稀释定容。 •(4)测定方法:将样品的盐酸提取液偶氮化后测定480nm的吸光度。标准曲线法定量。第二法:液相色谱法•(1)原理:水产品(鱼类及其制品、虾类及其制品)、肉类:试样用5%三氯乙酸提取,正已烷去除脂肪,三氯甲烷-正丁醇(1+1)液液萃取净化后,丹磺酰氯衍生,C18色谱柱分离,高效液相色谱-紫外检测器检测,内标法定量。酒类(葡萄酒、啤酒、黄酒等)、调味品(醋和酱油):试样用丹磺酰氯衍生,C18色谱柱分离,高效液相色谱-紫外检测器检测,内标法定量。•(2)范围:本标准规定了食品中色胺、β-苯乙胺、腐胺、尸胺、组胺、章鱼胺、酪胺、亚精胺和精胺含量的测定方法。本标准适用于酒类(葡萄酒、啤酒、黄酒等)、调味品(醋和酱油)、水产品(鱼类及其制品、虾类及其制品)、肉类中生物胺的测定。•(3)检出限:20mg/kg试样制备•4.1试样制备•取水产品及肉类样品的可食部分约500g,充分匀质,均分成两份装入洁净容器中,密封,于-20℃保存。•4.2提取•准确称取已经绞碎或匀浆后的水产品、肉类10g(精确至0.01g),置于100mL具塞锥形瓶中,加入500μL内标使用液 (1.0mg/mL)与样品充分混匀,加入20mL5%三氯乙酸溶液和振荡提取30min,转移至50mL具塞离心管中,5000r/min离心10min,转移上清液至50mL容量瓶中,残渣用20mL5%三氯乙酸溶液再提取一次,合并上清液,用5%三氯乙酸稀释至刻度,待净化。净化及萃取•4.3净化•4.3.1除脂肪:移取上述试样提取液10mL于25mL具塞试管中,加入0.5g氯化钠涡旋振荡至氯化钠完全溶解后加入10mL正己烷,涡旋振荡5min,静置分层后弃去上层有机相,下层试样溶液加入10mL正己烷再除脂一次。•4.3.2萃取:移取5mL上述除脂肪后的试样溶液于10mL具塞离心管中,用5mol/L氢氧化钠溶液(几滴)调节pH至12.0左右。加入5mL的正丁醇/三氯甲烷(1+1)混合溶液,涡旋振荡5min,5000r/min离心5min,转移上层有机相于另一个10mL具塞离心管中,下层样液再萃取一次,合并萃取液,用正丁醇/三氯甲烷(1+1)稀释至刻度。取5mL萃取液加入200μL盐酸(1mol/L),混匀后40℃水浴下氮气吹干,加入1mL盐酸(0.1mol/L)涡旋振荡,使残留物完全溶解,待衍生。衍生及标准曲线制作•4.4衍生•4.4.1试样的衍生:在上述待衍生的试样溶液中依次加入1mL饱和碳酸氢钠溶液、100μL氢氧化钠溶液(1mol/L)、1mL衍生试剂,涡旋混匀1min后置于60℃恒温水浴中衍生15min,取出,分加入100μL谷氨酸钠溶液,振荡混匀,60℃恒温反应15min。取出,冷却至室温,于每个离心管中加入1mL水,涡旋混合1min,40℃水浴下氮吹除去丙酮(约1mL),加入0.5g氯化钠涡旋振荡至氯化钠完全溶解后加入5mL乙醚,涡旋振荡2min,静置分层后,吸出上层有机相(乙醚层),再萃取一次,合并乙醚萃取液,40℃水浴下氮气吹干。加入1mL乙腈涡旋振荡使残留物完溶解,0.22μm滤膜针头滤器过滤于进样小瓶,待测定。•4.4.2标准的衍生:分别移取1mL生物胺标准系列溶液,置于10mL具塞试管中,依次加入250μL内标使用液(100mg/L),以下操作同试样的衍生步骤。衍生及标准曲线制作•5.测定方法•色谱柱为C18柱(柱长250mm,柱内径4.6mm,柱填料粒径5μm),紫外检测波长254nm,进样量20μL,柱温35,℃流动相 A为90%乙腈/10%(含0.1%乙酸的0.01mol/L乙酸铵溶液),流动相 B为10%乙腈/90%(含0.1%乙酸的0.01mol/L乙酸铵溶液),流速0.8mL/min。将试样的衍生溶液注入高效液相色谱仪中,测得峰面积,以保留时间定性。根据标准曲线得到待测液中各目标化合物的浓度。•5.2标准曲线的制作•将20μL系列混合标准工作液的衍生液分别注入高效液相色谱仪,测得目标化合物的峰面积,以系列混合标准工作液的浓度为横坐标,以目标化合物的峰面积与内标的峰面积的比值为纵坐标,绘制标准曲线。其他测定法:高效液相色谱-紫外检测法•1)原理:高效液相色谱法由于其具有分析速度快、柱效高、检测灵敏度高、定量分析 准确等特点, 基本取代了其他方法, 是目前生物胺含量分析测定的主要手段。衍生方法主要有柱前衍生和柱后衍生, 常用的衍生化试剂有邻苯二甲醛、丹磺酰氯、偶氮试剂和苯甲酰氯等, 邻苯二甲醛存在衍生物不稳定等不足, 苯甲酰氯不溶于水, 衍生效果差, 偶氮显色法容易受到基质中杂质的干扰, 影响测定结果。丹磺酰氯具有较理想的衍生效果和稳定时间。而有些标准中用盐溶液作为流动相, 会对分析柱造成损害。对高效液相色谱法测定水产品(鲭鱼)中组胺含量的试验方法进行改进,建立一种提取方法简单、衍生物稳定、操作方便的检测方法。•(2)方法说明:适用于鱼和虾等具有有毒生物胺的水产品。•本方法的检出限为50mg/kg。•(3)样品前处理:称取2 g(精确至 0.001 g)已粉碎的样品于 50 mL刻度离心管中, 用移液枪加入10 mL的0.4mol/L 高氯酸溶液,涡旋提取2 min,上离心机于4000 r/min 离心5 min, 将上清液移入25 mL棕色容量瓶中。在下层的残渣中再加入10 mL的0.4 mol/L的高氯酸溶液, 涡旋提取2min,离心后转移上清液于容量瓶中,用0.4mol/L 高氯酸溶液将其定容至刻度。其他测定法:高效液相色谱-紫外检测法•样品衍生:量取1 mL样品溶液于5 mL离心管中, 依次加入2 moL/L氢氧化钠溶液100μL、饱和碳酸氢钠溶液300μL和10 mg/mL丹磺酰氯溶液2 mL, 盖紧盖子, 在40℃下避光反应40min, 每5min 振荡1次。反应完毕后, 加入浓氨水100μL 终止衍生化反应, 放置 30 min。用甲醇将其定容至刻度5 mL, 混合均匀, 过0.45μm的有机滤膜, 待测。•(4)测定方法:色谱柱: Waters C18柱(150 mm×4.6mm, 5μm); 流速:1.0mL/min; 柱温: 35 ℃; 进样量: 20μL; 检测器: 紫外检测器; 检测波长: 254 nm; 流动相: A: 甲醇, B: 水; 梯度洗脱。的测定法无机砷无机砷测定法1、液相色谱-原子荧光光谱法(GB5009.11-2014)2、液相色谱-电感耦合等离子体质谱法 (GB5009.11-2014)3、液相-原子荧光法(参考文献:卢亭,夏慧丽,张今君.液相-原子荧光法测定水产品中无机砷方法的研究[J].食品研究与开发,2017,38(13):179-181)030201第一法:液相色谱-原子荧光光谱法•(1)原理:食品中无机砷经稀硝酸提取后,以液相色谱进行分离,分离后的目标化合物在酸性环境下与KBH4 反应,生成气态砷化合物,以原子荧光光谱仪进行测定。按保留时间定性,外标法定量。•(2)精密度:在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。检出限:0.03mg/kg•(3)样品前处理:•称取约1.0g水产动物湿样(准确至0.001g),置于50mL塑料离心管中,加人20mL0.15mol/L 硝酸溶液,放置过夜。于90C恒温箱中热浸提2.5h,每0.5h振摇1mm。提取完毕,取出冷却至室温, 8000 r/min离心15 min。取5 mL上清液置于离心管中,加人5 mL正己烷,振摇1 min后,8000 r/min离心15min,弃去上层正己烷。按此过程重复一次。吸取下层清液,经0.45μm有机滤膜过滤C18小柱净化后进样。按同一操作方法作空白试验。测定方法•(4)测定方法:原子荧光检测参考条件:负高压:320V;砷灯总电流:90mA;主电流/辅助电流:55/35;原子化方式:火焰原子化;原子化器 温度:中温。载液:0%盐酸溶液,流速4mL/min;还原剂:30g/L硼氢化钾溶液,流速4mL/min;载气流速: 400mL/min;辅助气流速:400mL/min。•标准溶液配制:亚砷酸盐[AS(H)]标准储备液(00mg/L,按AS计):准确称取三氧化二砷0.0132g,加 100g/L氢氧化钾溶液1mL和少量水溶解,转人100mL容量瓶中,加人适量盐酸调整其酸度近中性, 加水稀释至刻度。4°C保存,保存期一年。或购买经国家认证并授予标准物质证书的标准溶液物质。 25.4.2砷酸盐[As(V)]标准储备液(100mg/L,按AS计):准确称取砷酸二氢钾0.0240g,水溶解,转入100mL容量瓶中并用水稀释至刻度。4C保存,保存期一年。或购买经国家认证并授予标准物质证 书的标准溶液物质。标准曲线绘制•标准曲线绘制:取7支10mL容量瓶,分别准确加人1.00mg/L混合标准使用液0.00mL、0.050mL、0.10mL、0.20mL、0.30mL、0.50mL和l.0mL,加水稀释至刻度,此标准系列溶液的浓度分别为0.0ng/mL、 5.0ng/mL、10ng/mL、20ng/mL、30ng/mL、50ng/mLQl00ng/mL。•吸取标准系列溶液100mL注人液相色谱-原子荧光光谱联用仪进行分析,得到色谱图,以保留时间定性。以标准系列溶液中目标化合物的浓度为横坐标,色谱峰面积为纵坐标,绘制标准曲线。第二法:液相色谱-电感耦合等离子体质谱法•(1)原理:食品中无机砷经稀硝酸提取后,以液相色谱进行分离,分离后的目标化合物经过雾化由载气送人 ICP炬焰中,经过蒸发、解离、原子化、电离等过程,大部分转化为带正电荷的正离子,经离子采集系统进入质谱仪,质谱仪根据质荷比进行分离测定。以保留时间定性和质荷比定性,外标法定量。•(2) 精密度:在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。检出限:0.06mg/kg测定方法•(3)样品前处理:•称取约1.0g水产动物湿样(准确至0.001g),置于50mL塑料离心管中,加人20mL0.15mol/L硝酸溶液,放置过夜。于90C恒温箱中热浸提2.5h,每0.5h振摇1mm。提取完毕,取出冷却至室温, 8000r/min离心15min。取5mL上清液置于离心管中,加人5mL正己烷,振摇1min后,8000r/min离心15min,弃去上层正己烷。按此过程重复一次。吸取下层清液,经0.45μm有机滤膜过滤C18小柱净化后进样。按同一操作方法作空白试验。•(4)测定方法: •电感耦合等离子体质谱仪参考条件:RF入射功率1550W;载气为高纯氩气;载气流速0.85L/min;补偿气0.15L/min。泵速0.3rps;检测质量数 m/z=75(As),m/z=35(Cl)。•吸取试样溶液50μL注人液相色谱-电感耦合等离子质谱联用仪,得到色谱图,以保留时间定性。 根据标准曲线得到试样溶液中As(I)与As(V)含量,As(I)与As(V)含量的加和为总无机砷含量, 平行测定次数不少于两次。其他测定法:液相-原子荧光法•(1)原理:动物性水产品中无机砷经稀硝酸超声提取后,以液相色谱进行分离,分离后的目标化合物在酸性环境下与 KBH4反应,生成气态砷化合物,以原子荧光光谱仪进行测定。按保留时间定性,外标法定量。•(2)方法说明:该法具有操作便捷、干扰少、准确度好等优点,适用于批量水产品种无机砷的测定。检出限为0.01 mg/kg。•(3)样品前处理:准确称取经粉碎过 80目筛的样品1.0 g于50mL离心管中,加0.15 mol/L 的硝酸20.0mL旋涡混匀后置于超声波清洗器中提取,以8000r/min离心15min后取上清液5mL于10mL离心管中,加入5mL正己烷,振摇1min后,8000r/min离心15 min,弃去上层正己烷。按此过程重复一次,吸取下层清液,经0.45μm水相滤膜过滤及C18小柱净化后进样,同时做试剂空白试验。•(4)测定方法:色谱柱:阴离子交换色谱柱(HAMILTONPRP-X100 10μm 250mm×4.1 mm)。梯度洗脱:流动相 A:1mmol/L 磷酸氢二铵溶液(pH6.0);流动相 B:20mmol/L 磷酸氢 二铵溶液(pH6.0)。流动相、光谱等参数条件同国家标准GB 5009.11-2014《食品安全国家标准食品中总砷及无机砷的测定》的测定法甲基汞甲基汞测定法1、液相色谱-原子荧光光谱联用方法(GB5009.17-2014)2、气相色谱法(参考文献:刘素琴,牟晓崟.气相色谱法测定水产品中痕量甲基汞的方法研究[J].中国卫生检验杂志,2015,25(03):344-347)0201第一法:液相色谱-原子荧光光谱联用方法•(1)原理:食品中甲基汞经超声波辅助5 mol/L盐酸溶液提取后,使用反相色谱柱分离,色谱流出液进人在线紫外消解系统,在紫外光照射下与强氧化剂过硫酸钾反应,甲基汞转变为无机汞。酸性环境下,无机汞与硼氢化钾在线反应生成汞蒸气,由原子荧光光谱仪测定。由保留时间定性,外标法峰面积定量。•(2)方法说明:。检出限为0.038微克每毫升。•(3)样品前处理:•称取样品0.50 g~2.0 g (精确至0.001 g),置于15 mL塑料离心管中,加人10 mL的盐酸溶液 (5 mol/L),放置过夜。室温下超声水浴提取60 min,期间振摇数次。4 ℃下以8 000 r/min转速离心 15 min。准确吸取2.0mL清液至5 mL容量瓶或刻度试管中,逐滴加人氢氧化钠溶液(6mol/L),使样液pH为2~7。加人0.1 mL的L-半胱氨酸溶液(10 g/L),最后用水定容至刻度。0.45μm有机系滤膜过滤,待测。同时做空白试验。 测定方法•标准液的配制:氯化汞标准储备液(200μg/mL,以Hg计):准确称取0.0270g氯化汞,用0.5g/L重铬酸钾的硝酸溶液溶解,并稀释、定容至100mL。于4t冰箱中避光保存,可保存两年。甲基汞标准储备液(200μg/mL,以Hg计):准确称取0.0250g氯化甲基汞,加少量甲醇溶解, 用甲醇溶液(1+1)稀释和定容至100mL。于4t冰箱中避光保存,可保存两年。混合标准使用液(1.00μg/mL,以Hg计):准确移取0.50mL甲基汞标准储备液和0.50mL氯 化汞标准储备液,置于100mL容量瓶中,以流动相稀释至刻度,摇匀。此混合标准使用液中,两种汞化合物的浓度均为1.00μg/mL。现用现配。•标准曲线制作:取5支10mL容量瓶,分别准确加人混合标准使用液(1.00μg/mL)0.00mL、0.010mL、 0.020mL、0.040mL、0.060mL和0.10mL,用流动相稀释至刻度。此标准系列溶液的浓度分别为 0.0ng/mL、1.0ng/mL、2.0ng/mL、4.0ng/mL、6.0ng/mL和 10.0ng/mL。吸取标准系列溶液 100μL进样,以标准系列溶液中目标化合物的浓度为横坐标,以色谱峰面积为纵坐标,绘制标准曲线。•试样溶液的测定:将试样溶液100μL注人液相色谱-原子荧光光谱联用仪中,得到色谱图,以保留时间定性。以外标法峰面积定量。平行测定次数不少于两次。其他测定法:气相色谱法•(1)原理:建立一种快速、简便、灵敏的气相分析检测方法测定水产品中痕量甲基汞的含量,为水产品中痕量甲基汞残留量的测定方法改进提供新的方案。 •(2)方法说明:本实验提供的操作方法简便、灵敏,色谱峰分离度良好,峰形尖锐,重现性良好,系统适用性良好,且实验对环境污染较小,对实验人员的健康危害较小。•检出限为0.081 mg/kg。•(3)样品前处理:样品处理取匀浆处理后的样品约1 g,精密称定,置乳钵中,加入2. 0 g氯化钠进行研磨分散,再加入2mol/L的盐酸溶液 ml,继续研磨至液状,转移至离心管中,用8 ml盐酸溶液洗涤研钵,洗涤液转移至同一离心管中,匀速振摇60 min后离心过滤,滤液转入离心管中,精密加入二氯甲烷4ml,振荡萃取15 min,静置分层,吸取上层清液,经0.22μm滤膜过滤,即得供试品溶液。•4)测定方法:色谱条件 色谱柱: Agilent DB-1701 柱 ( 30 m×0.32 mm,1.00μm) ;检测器:电子捕获检测器;柱流量: 1.11ml/min; 分流比: 1:1; 进样量:1.0μl;检测器温度: 300 ℃; 进样口温度: 240 ℃; 柱温:初始温度140 ℃,保持 5.5 min,以5 ℃ /min 降温 至105 ℃,保持9 min,再以20 ℃ /min 升温至140 ℃,保持9 min。THANKS
 高中化学《第二节富集在海水中的元素----氯》PPT课件
高中化学《第二节富集在海水中的元素----氯》PPT课件 奇妙的化学振荡反应PPT课件下载
奇妙的化学振荡反应PPT课件下载 初中化学分子的课件PPT下载(共22页)
初中化学分子的课件PPT下载(共22页) 高一化学必修一氧化还原反应PPT课件下载
高一化学必修一氧化还原反应PPT课件下载 常见的金属材料PPT课件第1课时
常见的金属材料PPT课件第1课时 酶的化学结构PPT课件下载(共115页)
酶的化学结构PPT课件下载(共115页)