






《化学药品杂质研究基本思路及控制方法技PPT课件下载》是由用户上传到老师板报网,本为文库资料,大小为1.02 MB,总共有65页,格式为ppt。授权方式为VIP用户下载,成为老师板报网VIP用户马上下载此课件。文件完整,下载后可编辑修改。
- 文库资料
- 65页
- 1.02 MB
- VIP模板
- ppt
- 数字产品不支持退货
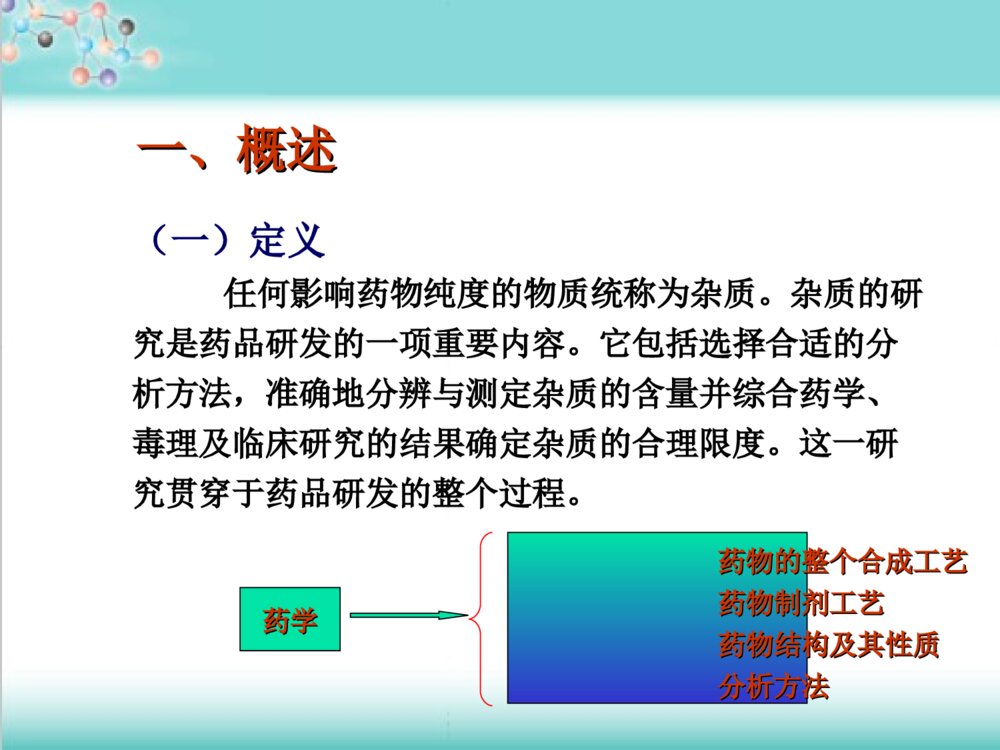
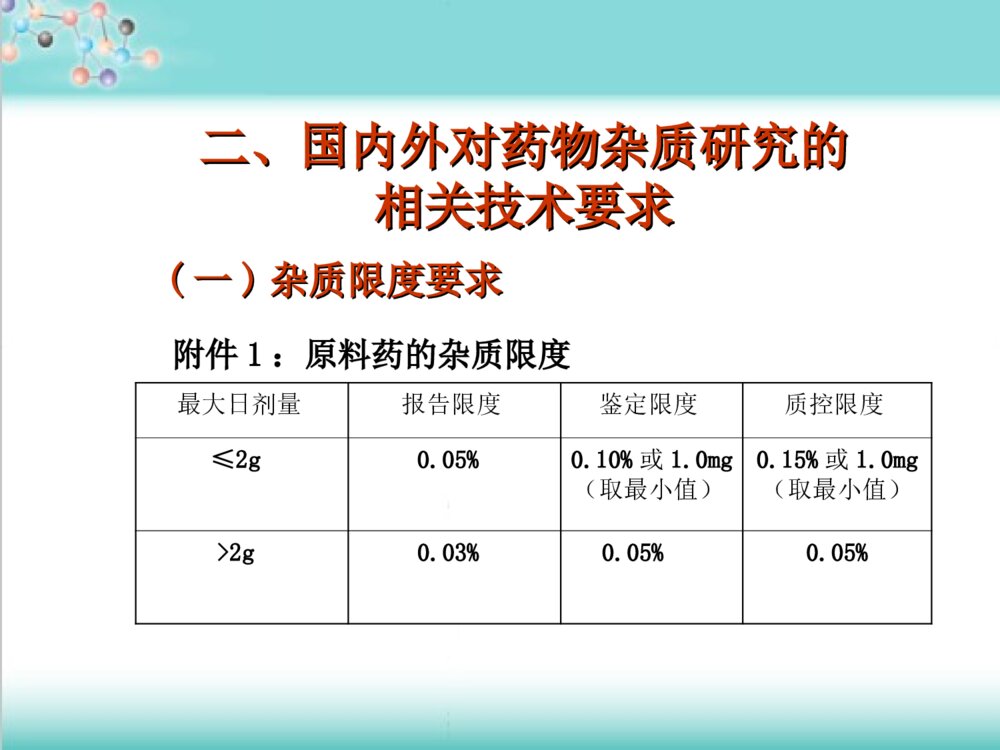
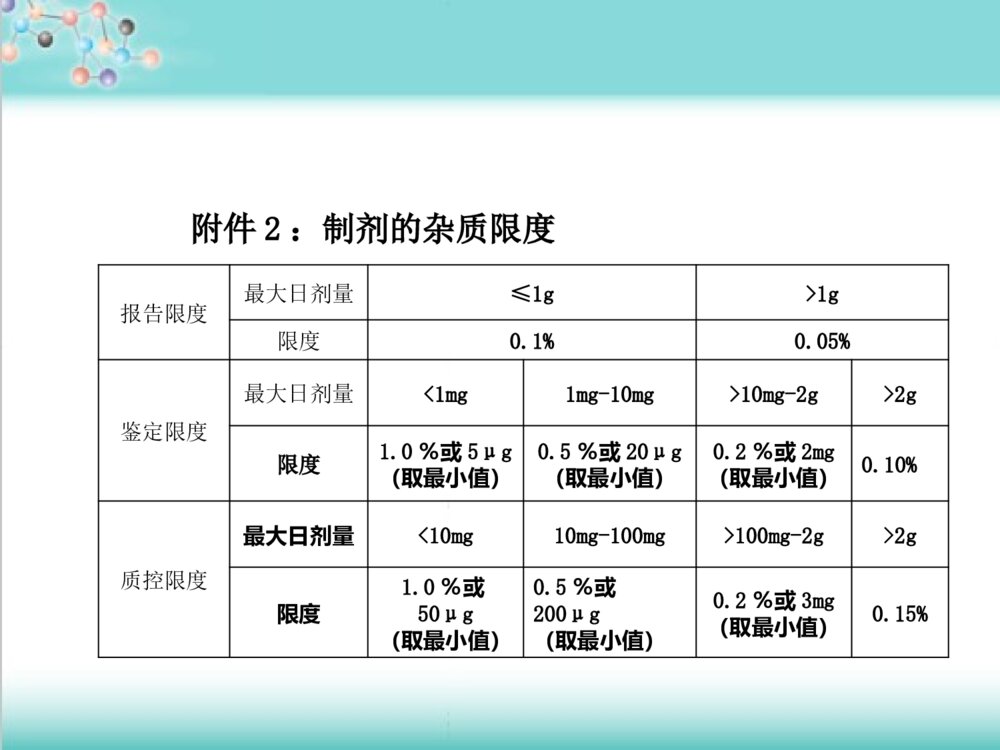
药物杂质研究基本思路药物杂质研究基本思路及控制方法及控制方法李眉1.国内外对药物杂质研究的相关技术要求2.杂质来源和控制有机杂质无机杂质残留溶剂主要内容主要内容ICH(InternationalConferenceonHarmonizationofTechnicalRequirementsforRegistrationofPharmaceuticalsforHumanUse)美国药典(USP)英国药典(BP)欧洲药典(EP)中国药典(ChP)《《化学药物杂质研究的技术指导原则化学药物杂质研究的技术指导原则》》相关内容的基础上,结合我国药物研发的特点,通过分析、研究与药物的安全性、有效性及质量可控性之间的内在关系而制定的一、概述一、概述(一)定义任何影响药物纯度的物质统称为杂质。杂质的研究是药品研发的一项重要内容。它包括选择合适的分析方法,准确地分辨与测定杂质的含量并综合药学、毒理及临床研究的结果确定杂质的合理限度。这一研究贯穿于药品研发的整个过程。药学药学药物的整个合成工艺药物的整个合成工艺药物制剂工艺药物制剂工艺药物结构及其性质药物结构及其性质分析方法分析方法(二)杂质的分类按理化性质按理化性质按照其来源按照其来源按照其毒性按照其毒性按化学结构按化学结构有机杂质、无机杂质、残留溶剂工艺杂质(包括合成中未反应完全的反应物、试剂、中间体、副产物等)降解产物从反应物及试剂中混入的杂质等。毒性杂质、普通杂质如甾体、生物碱、几何异构体、光学异构体聚合物等。有机杂质:包括工艺中引入的杂质和降解产物等,由于这类杂质的化学结构一般与活性成分类似或具渊源关系,故通常又可称之为有关物质。无机杂质:是指在原料药及制剂生产或传递过程中产生的杂质,这些杂质通常是已知的,主要包括:反应试剂、配位体、催化剂、重金属、其它残留的金属、无机盐、助滤剂、活性炭等。残留溶剂:在合成、纯化、制剂过程中残留所使用的溶剂,这些溶剂通常是已知的。二、国内外对药物杂质研究的二、国内外对药物杂质研究的相关技术要求相关技术要求最大日剂量报告限度鉴定限度质控限度≤2g0.05%0.10%或1.0mg(取最小值)0.15%或1.0mg(取最小值)>2g0.03%0.05%0.05%附件1:原料药的杂质限度((一一))杂质限度要求杂质限度要求报告限度最大日剂量≤1g>1g限度0.1%0.05%鉴定限度最大日剂量<1mg1mg-10mg>10mg-2g>2g限度1.0%或5μg(取最小值)0.5%或20μg(取最小值)0.2%或2mg(取最小值)0.10%质控限度最大日剂量<10mg10mg-100mg>100mg-2g>2g限度1.0%或50μg(取最小值)0.5%或200μg(取最小值)0.2%或3mg(取最小值)0.15%附件2:制剂的杂质限度报告限度(ReportingThreshold):超出此限度的杂质均应在检测报告中报告,并应报告具体的检测数据。鉴定限度(IdentificationThreshold):超出此限度的杂质均应进行定性分析,确定其化学结构。质控限度(QualificationThreshold):质量标准中一般允许的杂质限度,如制订的限度高于此限度,则应有充分的依据。11、有机杂质的限度确定、有机杂质的限度确定质量标准中对有机杂质的限度规定应包括:每一个已知杂质、未知杂质及总杂质。共存的异构体和抗生素的多组分一般不作为杂质进行控制,必要时作为共存物质在质量标准中规定其比例。单一的对映体药物,其对映异构体应作为杂质控制.由于创新药物与仿制药情况不同,在确定杂质限度时路有所区别。(1)创新药物由于在创新药物的研究过程中进行药理毒理临床研究超出了附件1或2的质控限度,仍可认为该杂质的含量已经通过了安全性的验证。在此前提之下,如果该杂质的含量同时也在正常的制备工艺所允许的限度范围内,那么根据试验样品中杂质的含量所确定的限度可认为是合理的。故新产品应在上市后继续监测不良反应:如与杂质有关,则应分析原因,设法降低杂质含量,这样制订出来的杂质限度才能保证产品的安全性。如某杂质同时也是该药物在动物或人体中的主要代谢产物,则对该杂质可不考虑其安全性,但需制订合理的限度。与药物性质和临床应用相关联。盐酸甲砜霉素甘氨酸酯NHCOCHCl2CHOHCHCH2OHNHCOCHCl2CH3SO2CHOHCHNHCOCHCl2CH3SO2CH2OCOCH2ClClCH2COClDMFPy12CHOHCHCH3SO2CH2OCOCH2N4(CH2)6Cl(CH2)6N4/CH3CNHClC2H5OH+C2H5OC2H5CHOHCHNHCOCHCl2CH3SO2CH2OCOCH2NH2.HClCHOHCHCH2OHNHCOCHCl2CH3SO21福莫司汀CH3CH2OCH3CH2OPOCHCH3NHCONNOCH2CH2ClCH3COCl+CH3COPO(OCH2CH3)2CH3CNPOOH(OCH2CH3)2NH2OH.HClPO(OCH2CH3)21.Zn/HCOOH2.HCl/CH3OHCH3CHNH2CH3CH2OCH3CH2OPOCHCH3NHCONHCH2CH2ClClCH2CH2NCONaNO2/HCOOHP(OCH2CH3)3(I)(II)(III)(IV)(2)仿制已有国家标准的药品可以根据已有的标准制订相应的杂质限度。1、该标准中未规定杂质的限度:应与已上市同品种药品进行全面的质量对比研究杂质的种类与含量稳定性考察2、难以获得已上市同品种的标准,但有相同原料药的其它剂型上市,则在制订杂质限度时路参考此上市产品质量标准,对杂质进行控制。3、工艺或处方的不同杂质的限度:新杂质的含量高于附件1或2规定的合理限度在研产品的杂质含量明显高于已上市的同品种产品的杂质实测值。优化产品的处方与制备工艺安全性研究。(3)其它新药改变给药途径的制剂,其杂质限度的确定参照创新药物的要求进行。对于其它类别的新药,如果能够获得已上市的对照样品,则可按照仿制已有标准的药品的研究思路,在详细的质量对比研究的基础上,确定杂质的限度。如果不能获得对照样品,则应参照创新药物的要求确定杂质限度,或通过详细的安全性试验来证明已有的杂质限度是安全的。22、无机杂质的限度确定、无机杂质的限度确定原则:无机杂质的限度主要根据该杂质的毒性、对药品本身质量(如稳定性)的影响及各批次产品的实测结果而定。各国药典收载的质量标准及我国已批准上市产品的注册标准中对于我们确定在研产品的无机杂质限度具有重要的参考价值。给药途径、适应症、剂量等选择合适的参考标准,确定合理的限度。重金属0.001%铁盐0.003%氯化物0.014%硫酸盐0.04%-0.1%砷盐0.0002-0.0005%33、残留溶剂限度确定、残留溶剂限度确定药物中常见残留溶剂及其限度溶剂名称PDE值(mg/天)限度(%)第一类溶剂(应避免使用)苯0.020.0002四氯化碳0.040.00041,2-二氯乙烷0.050.00051,1-二氯乙烯0.080.00081,1,1-三氯乙烷15.00.15溶剂名称PDE值(mg/天)限度(%)第二类溶剂(应该限制使用)乙腈4.10.041氯苯3.60.036氯仿0.60.006环己烷38.80.388N,N-二甲氧基甲酰胺8.80.0881,4-二氧六环3.80.038正己烷2.90.029甲醇30.00.3四氢呋喃7.20.072甲苯8.90.089二氯甲烷6.00.06溶剂名称PDE值(mg/天)限度(%)第三类溶剂(GMP或其他质量要求限制使用)叔丁基甲基醚50.00.5异丙基苯50.00.5二甲亚砜50.00.5乙醇50.00.5乙酸乙酯50.00.5乙醚50.00.5甲酸乙酯50.00.5甲酸50.00.5正庚烷50.00.5乙酸异丁酯50.00.5乙酸异丙酯50.00.544、临床研究申请与上市生产申请、临床研究申请与上市生产申请阶段的杂质研究阶段的杂质研究申报临床研究:1、申报临床研究前,应对已有批次产品的杂质进行比较,根据安全性研究用样品的杂质含量情况来证明临床研究用药品是安全的。2、可在临床研究期间对杂质分析方法进行完善。3、对于创新药物路对杂质的限度做初步的规定。申报生产研究:1、产生了新的杂质2、已有杂质的含量超出限度根据附件1或2来判断该杂质的含量是否合理,如不合理,则应参照决策树来考虑下一步的研究工作。(二)杂质检测技术要求(二)杂质检测技术要求11、杂质分析方法、杂质分析方法分析方法的选择直接关系到杂质测定结果的专属性与准确性,因此,在进行杂质研究时首要问题是选择合适的杂质分析方法。(1)有机杂质的分析方法化学法、光谱法、色谱法等,随着分离、检测技术的发展与更新,高效、快速的分离技术与灵敏、稳定、准确、适用的检测手段相结合。目前普遍采用的杂质检测方法:高效液相色谱法薄层色谱法、气相色谱法毛细管电泳法应根据药物及杂质的理化性质、化学结构、杂质的控制要求等确定适宜的检测方法。由于各种分析方法均具有一定的局限性,因此在进行杂质分析时,应注意不同原理的分析方法间的相互补充与验证,如HPLC与TLC及HPLC与CE的互相补充,反相HPLC系统与正相HPLC系统的相互补充,HPLC不同检测器检测结果的相互补充等。NH2COOCH3NH2COOCH3BrBrNH2CONHNH2BrBrNH2CONHNHBrBrSOOOCH3NH2BrBrCH=NOHNH2BrBrCH2-NHOHOHNH2(2)无机杂质的分析方法无机杂质的产生主要与生产工艺过程有关。由于许多无机杂质直接影响药品的稳定性,并可反映生产工艺本身的情况,了解药品中无机杂质的情况对评价药品生产工艺的状况有重要意义。对于无机杂质,各国药典都收载了经典、简便而又行之有效的检测方法。对于成熟生产工艺的仿制路根据实际情况,采用药典收载的方法进行质量考察及控制。对于采用新生产工艺生产的新药,鼓励采用离子色谱法及电感耦合等离子发射光谱-质谱(ICP-MS)等分析技术,对产品中可能存在的各类无机杂质进行定性、定量分析,以便对其生产工艺进行合理评价,并为制定合理的质量标准提供依据。不挥发性无机杂质采用炽灼残渣法进行检测。某些金属阳离子杂质(银、铅、汞、铜、镉、铋、锑、锡、砷、锌、钴与镍等)笼统地用重金属限度检查法进行控制。因在药品生产中遇到铅的机会较多,且铅易积蓄中毒,故作为重金属的代表,以铅的限量表示重金属限度。对某种(些)特定金属离子或上述方法不能检测到的金属离子作限度要求,可采用专属性较强的原子吸收分光光度法或具有一定专属性的经典比色法(如采用药典已收载的铁盐、铵盐、硒等的检查法检测药品中微量铁盐、铵盐和硒等杂质)。虽然重金属检查法可同时检测砷,但因其毒性大,且易带入产品中,故需采用灵敏度高、专属性强的砷盐检查法进行专项考察和控制,各国药典收载的方法已历经多年验证,行之有效,应加以引用。(3)残留溶剂的检测方法在确定了需要进行残留量研究的溶剂后,需要通过方法学研究建立合理可行的检测方法。目前,常用的检测方法为气相色谱法(GasChromatography,GC),也有其他一些检测方法。三、杂质来源和控制三、杂质来源和控制(一)有机杂质来源和控制(一)有机杂质来源和控制工艺杂质反应物试剂中间体副产物降解产物混入的杂质反应物试剂工艺杂质NH2COOCH3NH2COOCH3BrBrSOOCH3NH2BrBrCH=NOHNH2CONHNHBrBrNH2BrBrCH2-NHOHNH2CONHNH2BrBrO反应物、试剂、中间体盐酸氨溴索合成NH2COOCH3NH2COOCH3BrNH2COOCH3BrNH2CONHNH2NHNH2BrNH2COOCH3NH2COOCH3BrBrNH2CONHNH2BrBr副产物异构体NH2BrBrCH2-NHOHCF3OONHHNOHHHHHCF3OONHHNOHHHH2OF3CCONHNH2CF3OONHHNOHHHHHOOOHHHHHHOOOHHOONHHNHOCF3F3CCONHNH2H2OOOO+OOOHHOOOHHHHHH不同工艺路线NH2CHONH2CHOBrBrNH2BrBrCH=NOHNH2BrBrCH2-NHOH桂哌齐特工艺研究NHH3COH3COH3COCHCHCOOH+SOCl2ClCH2COClEt3NinCH2Cl2NCOCH2ClpiperazineK2CO3inEtOHNCOH2CNNHH3COH3COH3COCHCHCOClEt3NCH2Cl2COOHCOOHCOOHCOOHH3COH3COH3COCHCHCONNH2CCNO.副产物CH3OCH=CHCOClCH3OCH3OCH3OCH=CHCOCH3OCH3ONNH异构体CH3OC=CNNCH2CCH3OCH3OCONHHCH3OCH=CHCOClCH3OCH3OO降解产物CH3OCH=CHNNCH2CH3OCOCH3OCH=CHCOClOCH3OCH3OCH3OCON不同工艺路线NHClCH2COClEt3NinCH2Cl2NCOCH2ClpiperazineK2CO3inEtOHNCOH2CNNHClCH2COClPPh3inCH3CNNCOH2CNNPPh3OHClClH3COH3COH3COCHCHONaOH,CH2Cl2COOHCOOHH3COH3COH3COCHCHCONNH2CCNO.COOHCOOH环丙沙星NFCOOHONHNNFCOOHOClNCOOHONHNNFCOOHONH2NHNCOOHONHNClNH2NHNCOOHOClNFONHN中间体NFCOOHOCl副产物NCOOHONHNCl副产物NFCOOHONH2NH副产物NH2NHNCOOHOCl降解产物NCOOHONHN降解产物NFONHNOORNNONH2OPOOHOHOHNNOONNONHH+NNONHCOOROHNNONHNNONHOHCOCOOPOOHOHOPOOEtOEtCOOHOHOPOOHOHNNONHOORC+NNONHOCOOHOPOOEtOEtOHOPOOHOHNNONHCONNONHCOHORNNONH2OHOPOOHOHOH有机杂质的定量方法有机杂质的检测一般多采用HPLC法,有时也采用TLC、GC等其它方法。如采用HPLC法,须采用峰面积法,①外标法(杂质对照品法)②加校正因子的主成分自身对照法③不加校正因子的主成分自身对照法④峰面积归一化法。①法定量比较准确②法应对校正因子进行严格测定,仅适用于已知杂质的控制③法的前提是假定杂质与主成分的响应因子基本相同。④法简便快捷,但因各杂质与主成分响应因子不一定相同、杂质量与主成分量不一定在同一线性范围内、仪器对微量杂质和常量主成分的积分精度及准确度不相同等因素,所以在质量标准中采用有一定的局限性。有关物质中包括已知杂质和未知杂质。已知杂质对主成分的相对响应因子在0.9-1.1范围内时,可以用主成分的自身对照法计算含量,超出0.9-1.1范围时,宜用杂质对照品法计算含量,也可用加校正因子的主成分自身对照法。理想的定量方法为已知杂质对照品法与未知杂质不加校正因子的主成分自身对照法两者的结合。研究人员可根据实际情况选用合适的定量方法。有机杂质的定量要求1、专属性为了考察在其他成分存在的条件下,采用的方法是否具有准确测定出待检测的有机杂质的能力。2、检测限通常有机杂质量较低,而每种有机杂质的检测灵敏度又各不相同,为了考察所采用的方法能否将残留的少量或微量的有机杂质检出。3、定量限为保证测定方法的准确度和精密度,需要进行定量限的研究。4、线性在配制对照品溶液时,对照品溶液浓度很难和规定的限度达到完全一致,当不一致时,需要通过标准曲线进行换算,这种换算的前提是有关物质的浓度与色谱峰面积直接成正比关系。5准确度为了保证检测结果能够代表产品的实际情况,建议进行方法的准确度考察。6耐用性为考察测定条件发生细小变动时,测定方法和结果是否仍准确可靠,建议进行耐用性考察。(二)无机杂质来源和控制工艺杂质反应物:盐试剂:含铅催化剂:镍、钯炭干燥剂:硫酸盐、氯化物、磷酸盐pH调节剂(两性药物)混入的杂质反应物试剂助滤剂、活性炭OOEtOOOOH3CH3CHNO3O165℃H3CH2NHOHOOOEtFe+HClH3COONNHClCHheatOOOEtOONH2OH3CH3COOEtOONO2OH3COBr(COCl)2OONNHNHOOH3CH3C+OONNHOH3COH3COOH3COH3CHCOONH4CHTBAlK2CO3OHNNOHNO2NNH-RNO2NO:NNNNNNH2ADCNNNNNNHREFB1B2NClNO2A硝化反应:68%硝酸+丙酸于125℃反应30min。B胺化反应:B1氯化,SOCl2+DMF于CH2Cl2中加热回流3.5hB2胺化,于-15℃滴加异丁胺与CH2Cl2溶液后,加热回流30min.C氢化反应:室温,RenyNi催化,50psi氢压力下氢化3h.D环合反应:甲酸中加热回流4h.E氧化反应:16%过氧乙酸+乙醇,于50℃反应24h.F胺化反应:二氯甲烷+28-30%氨水于0℃20min内滴加对甲苯磺酰氯的二氯甲烷溶液。详细的精制方法取咪喹莫特成品10g,加5N盐酸200ml,搅拌加热至80℃使溶解,过滤除去不溶物,滤液冷却,用二氯甲烷洗(50mlx2),加入活性炭少许,于80℃加热15min,滤去活性炭,滤液加连二亚硫酸钠3.3g,于80℃加热15min,冷却,过滤,固体溶于少量甲醇,加1N氢氧化钾甲醇溶液中和,滤出固体,用甲醇洗得6.6g咪喹莫特精制品。四、结论1.应注意对杂、质检测方法的选择与验证。2.应注意对研究过程中所有批次的样品中的杂质进行完整的记录,这些数据将是制订杂质限度的重要依据。3.应特别注意,在确定杂质的限度时,一定要综合考虑杂质的安全性、生产的可行性与产品的稳定性。4.在确定仿制药品的杂质限度时,应与已上市产品进行质量对比研究,以确保产品的安全性。
 《原子核外电子排布》化学九年级上第3章 构成物质的微粒ppt课件
《原子核外电子排布》化学九年级上第3章 构成物质的微粒ppt课件 《溶解度》溶液北京课改版九年级化学下册PPT课件
《溶解度》溶液北京课改版九年级化学下册PPT课件 化学用品安全培训PPT课件下载
化学用品安全培训PPT课件下载 《第三节 羧酸酯》化学PPT课件下载
《第三节 羧酸酯》化学PPT课件下载 化学探秘水世界PPT课件下载
化学探秘水世界PPT课件下载 《常见的酸和碱》初三九年级化学PPT课件
《常见的酸和碱》初三九年级化学PPT课件